|
Introduction
Protein microarrays are ideal reagents for high-throughput experiments that require very small amounts of analyte, and therefore can be used very efficiently to analyze many kinds of molecular interactions en masse including protein–nucleic acid and protein–small molecule interactions.
Although enormous progress has been made in the immobilization of DNA biomolecules onto different types of solid supports, the immobilization of proteins has been a particularly challenging task. Most of the available methods for the immobilization of proteins onto solid supports have traditionally relied on non-specific adsorption or on the reaction of naturally occurring chemical groups within proteins (mainly amines and carboxylic acids) with complementary reactive groups chemically introduced onto the solid support. In both cases, the corresponding proteins are attached to the surface in random orientations, which may cause the loss of the protein’s biological activity. Site-specific covalent immobilization, on the other hand, allows the proteins to be arranged in a definite, controlled fashion. This process requires the presence of two unique and mutually reactive groups on the protein and the support surface. The reaction between these two groups should be highly chemoselective, thus behaving like a molecular Velcro.
Protein Immobilization Using Expressed Protein Ligation
Our group has pioneered the use of expressed protein ligation (EPL) for the chemoselective attachment of biologically active proteins to produce protein microarrays [2]. Key to this approach was the use of recombinantly produced protein a-thioesters [3], which can be attached efficiently to surfaces containing N-terminal Cys residues through native chemical ligation (NCL) (Fig. 1). In this reaction, independently developed by Kent [4] and Tam [5], two fully unprotected polypeptides, one containing a C-terminal a-thioester group and the other a N-terminal Cys residue, react chemoselectively under neutral aqueous conditions to form a native peptide bond at the ligation site (Fig. 1a). We have recently used this process for the direct immobilization of several biologically active proteins onto modified glass surfaces through their C-termini (Fig. 1b) [2].
Although chemoselective ligation reactions such as a NCL are powerful tools for the site-specific and traceless immobilization of proteins through their N- or C-termini, they still require the use of relatively high concentrated solutions of the corresponding protein derivative. In the case of NCL, we have estimated that the minimal protein concentration required for an acceptable level of immobilization was ≈40 µM [2]. Other chemoselective ligation reactions, such as Staudinger ligation of “click” chemistry, have also been shown to have similar concentration requirements [6].
Protein Trans-Splicing: A Traceless Capture Ligand Approach for Protein Immobilization
The idea of using surfaces coated with reactive ligands for the covalent capture of proteins in order to allow selective covalent immobilizations under very diluted conditions has recently been reported by several groups (Fig. 2) [6]. In this approach, the protein to be immobilized is fused through its C- or N-terminus to a capture protein, usually an enzyme. The enzyme moiety then reacts selectively with the capture ligand immobilized onto the surface. This approach has been shown to work quite well with complex mixtures such as cellular lysates, where the concentration of the proteins to be immobilized are typically in the low µM range.
One of the main limitations of this approach, however, is that the capture protein remains attached to the surface after the immobilization process has taken place (Fig. 2). To address this problem, our group has recently developed a new traceless capture ligand approach for the selective immobilization of proteins to surfaces based on the protein trans-splicing process (Fig. 3) [7]. This process is similar to protein splicing [8] with the only difference being that the intein self-processing domain is split in two fragments (called N-intein and C-intein, respectively) [9].
In our approach, the C-intein fragment is covalently immobilized onto a glass surface through a PEGylated-peptide linker, while the N-intein fragment is fused to the C terminus of the protein to be attached to the surface (Fig. 3). When both intein fragments interact, they form an active intein domain, which ligates the protein of interest to the surface at the same time the split intein is spliced out into solution. Key to this approach was the use of the naturally split intein DnaE intein from Synechocystis sp. PCC6803. The C- and N intein fragments of this intein are able to self-assemble spontaneously (Kd = 0.1-0.2 µM), not requiring any refolding step [7, 10]. Moreover, the DnaE intein-mediated trans-splicing reaction is also very efficient under physiological-like conditions (t½ ≈ 4 h and trans-splicing yields ranging from 85% to almost quantitative) [7]. Using this split intein, we have successfully immobilized several proteins to chemically modified SiO2-based substrates (Fig. 3) [7].
Protein immobilization using protein trans-splicing is highly specific and efficient. It allows the use of protein mixtures and eliminates the need for the purification and/or reconcentration of the proteins prior to the immobilization step (Fig. 3). The required minimum protein concentration for efficient immobilization was estimated to be sub-micromolar [7]. More importantly, once the protein is immobilized to the surface, both intein fragments are spliced out into solution, providing a completely traceless method of attachment. All these features allow this methodology to be easily interfaced with cell-free protein expression systems with rapid access to the high-throughput production of protein chips and other types of biosensors.
Concluding Remarks
Immobilization of proteins onto solid supports is of great importance in numerous applications, including protein analysis, drug screening, and medical diagnostics, among others. The success of all these technologies relies on the immobilization technique employed to attach a protein to the corresponding surface. The recent introduction of protein trans-splicing makes it possible for the first time to interface a site-specific and traceless method for the immobilization of proteins with high-throughput expression systems thus allowing rapid access to high-quality protein chips.
References
[1] Muir, T.W., Annu. Rev. Biochem., 72, 249–89 (2003).
[2] Camarero, J.A., et al., J. Am. Chem. Soc., 126, 14730–1 (2004).
[3] Noren, C.J., et al., Angew. Chem. Int. Ed., 39, 451–6 (2000).
[4] Dawson, P.E., et al., Science, 266, 776–9 (1994).
[5] Tam, J.P., et al., Proc. Natl. Acad. Sci. USA, 92, 12485–9 (1995).
[6] Woo, Y.-H., et al., Curr. Nanoscience, 2, 93–103 (2006).
[7] Kwon, Y., et al., Angew. Chem. Int. Ed. , 45, 1726–9 (2006).
[8] Perler, F.B., et al., Curr. Opin. Biotechnol., 377–83 (2000).
[9] Perler, F.B., Trends Biochem. Sci., 24, 209–11 (1999).
[10] Shi, J., et al., J. Am. Chem. Soc., 127, 6198–206 (2005).
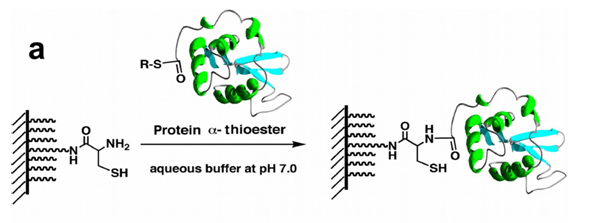
Figure 1. (a) General scheme for the chemoselective immobilization of protein a-thioesters onto N-terminal Cys-coated surfaces through NCL.

Figure 1. (b) Immobilization of fluorescent proteins EGFP (green) and DsRed (red) using NCL. The concentration of the proteins spotted onto the derivatized glass slide was ≈ 100 µM (≈1 mg/mL).
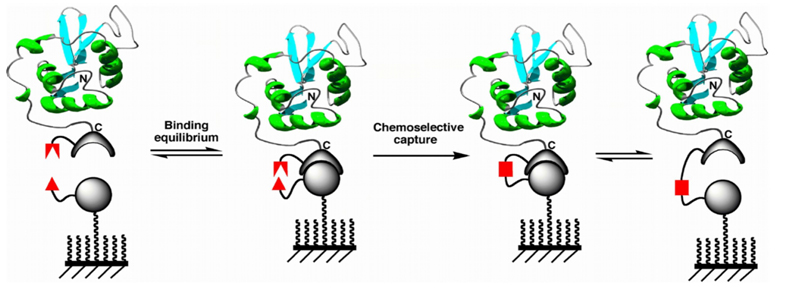
Figure 2. Protein immobilization through a capture ligand approach.
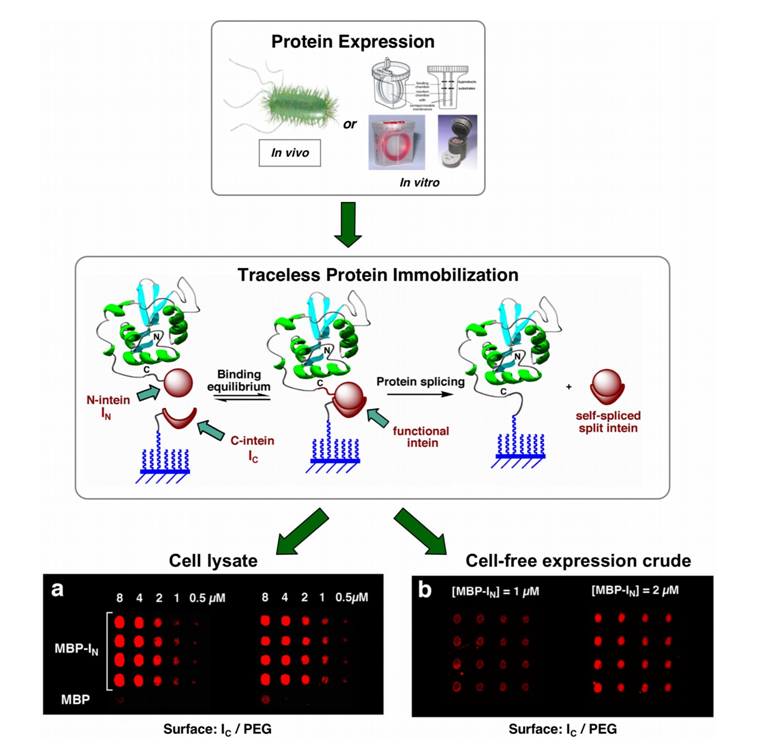
Figure 3. Site-specific and traceless immobilization of proteins onto solid supports through protein trans-splicing. Maltose binding protein (MPB) was directly immobilized without performing any purification or reconcentration step from a) soluble cellular fraction of E. coli cells overexpressing MBP-IN, and b) MBP-IN expressed in vitro using a cell-free expression system. MBP was detected by immunofluorescence. MBP-IN concentration on the different samples spotted was estimated by western blot analysis.
Dr. Julio A. Camarero is Associate Professor at the Department of Pharmaceutical Sciences in the University of Southern California since 2008. He studied chemistry at the University of Barcelona, received Masters Degree in 1992 and completed his PhD Thesis in 1996. Then he joined the group of Professor Tom W. Muir at The Rockefeller University as a Burroughs Wellcome Fellow where he contributed to the development of new chemo selective ligation techniques for the chemical engineering of proteins to study bacterial transcription. In 2000, he moved to the Lawrence Livermore National Laboratory as a Distinguished Lawrence Fellow where he became staff scientist and head of laboratory in 2003. He finally joined the School of Pharmacy at the University of Southern California in 2008 as Associate Professor. His current research interests are focused in the development of new bioorganic approaches using protein splicing and synthetic protein chemistry for studying biological processes involved in bacterial pathogenicity and cancer and how can be modulated or inhibited by highly constrained cyclic peptides. Dr. Camarero has authored over 40 peer-reviewed publications and four invited book chapters.
For article feedback, contact Dr. Camarero at
jcamarer@pharmacy.usc.edu
 |
 |
|
|
|
