|
High-throughput assays are indispensable for
comprehensive functional proteome research. The
development of these techniques has been driven by
the complete mapping of many genomes, including the
human. Of great importance for achieving this goal
is the development of new protein capture tools for
the detection and identification of specific
proteins. New capture reagents should be stable to
thermal and proteolytic degradation, have high
affinity, easy to produce and present low
cross-reactivity.
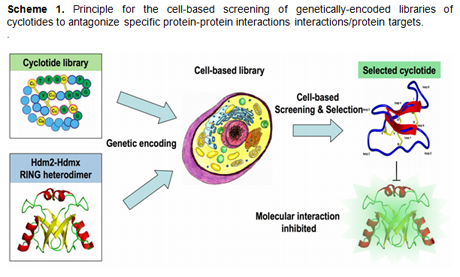 To
achieve this objective we are using a cyclotide-based
molecular scaffold for generating molecular
libraries that will be screened and selected in vivo
for potential antagonists against specific protein
targets or protein interactions. In this innovative
approach, we are using a cell-based library (E. coli
cell libraries) where every single cell will express
a different cyclotide, in what we could call a
single cell-single compound approach. These
compounds are then screened and selected for their
ability to inhibit a particular interaction inside
the bacterial cell using a genetically-encoded
reporter in combination with high throughput flow
cytometry to identify bacteria encoding specific
cyclotide-based antagonists (Scheme 1). Cyclotides,
a novel type of peptide-based protein-capture
reagent. Cyclotides are fascinating micro-proteins
present in plants from the Violaceae, Rubiaceae and
also Cucurbitacea and featuring various biological
actions such as toxic, inhibitory, anti-microbial,
insecticidal, cytotoxic, anti-HIV or hormone-like
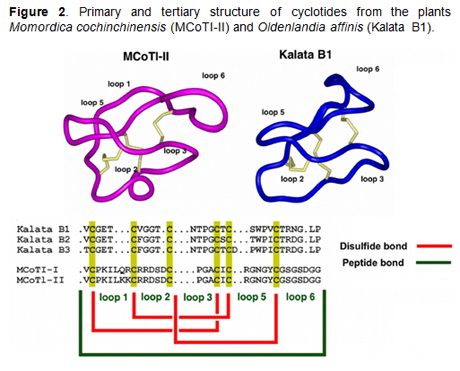
activity [1, 2]. They share a unique head-to-tail
circular knotted topology of three disulfide
bridges, with one disulfide penetrating through a
macrocycle formed by the two other disulfides and
inter-connecting peptide backbones, forming what is
called a cystine knot topology (Fig. 1 Cyclotides
belong to the family of knottins, a group of
microproteins that also includes conotoxins (389
sequences) and spider toxins (257 sequences).
Basically, cyclotides are knottins with a
head-to-tail circular topology. These micro-proteins
can be considered as natural combinatorial peptide
libraries structurally constrained by the cystine-knot
scaffold [2] and head-to-tail cyclization but in
which hypermutation of essentially all residues are
permitted with the exception of the strictly
conserved cysteines of the knot. The main features
of cyclotides and knottins in general are therefore
a remarkable stability due to the cystine knot, a
small size making them readily accessible to
chemical synthesis, and an excellent tolerance to
sequence variations. Cyclotides and knottins thus
appear as promising leads or frameworks for peptide
drug design [3, 4] and as extremely versatile and
stable protein capture reagents. To
achieve this objective we are using a cyclotide-based
molecular scaffold for generating molecular
libraries that will be screened and selected in vivo
for potential antagonists against specific protein
targets or protein interactions. In this innovative
approach, we are using a cell-based library (E. coli
cell libraries) where every single cell will express
a different cyclotide, in what we could call a
single cell-single compound approach. These
compounds are then screened and selected for their
ability to inhibit a particular interaction inside
the bacterial cell using a genetically-encoded
reporter in combination with high throughput flow
cytometry to identify bacteria encoding specific
cyclotide-based antagonists (Scheme 1). Cyclotides,
a novel type of peptide-based protein-capture
reagent. Cyclotides are fascinating micro-proteins
present in plants from the Violaceae, Rubiaceae and
also Cucurbitacea and featuring various biological
actions such as toxic, inhibitory, anti-microbial,
insecticidal, cytotoxic, anti-HIV or hormone-like
activity [1, 2]. They share a unique head-to-tail
circular knotted topology of three disulfide
bridges, with one disulfide penetrating through a
macrocycle formed by the two other disulfides and
inter-connecting peptide backbones, forming what is
called a cystine knot topology (Fig. 1 Cyclotides
belong to the family of knottins, a group of
microproteins that also includes conotoxins (389
sequences) and spider toxins (257 sequences).
Basically, cyclotides are knottins with a
head-to-tail circular topology. These micro-proteins
can be considered as natural combinatorial peptide
libraries structurally constrained by the cystine-knot
scaffold [2] and head-to-tail cyclization but in
which hypermutation of essentially all residues are
permitted with the exception of the strictly
conserved cysteines of the knot. The main features
of cyclotides and knottins in general are therefore
a remarkable stability due to the cystine knot, a
small size making them readily accessible to
chemical synthesis, and an excellent tolerance to
sequence variations. Cyclotides and knottins thus
appear as promising leads or frameworks for peptide
drug design [3, 4] and as extremely versatile and
stable protein capture reagents.
 Cyclotides
are ribosomally produced in plants from precursors
that comprise between one and three cyclotide
domains, however the mechanism of excision of the
cyclotide domains and ligation of the free N- and
C-termini to produce the circular peptides has not
been elucidated. Our group has recently developed
and successfully used a bio-mimetic approach for the
biosynthesis of folded cyclotides inside cells by
making use of modified protein splicing units (Fig.
2). Our important finding makes possible the
generation of large libraries of cyclotides (≈109)
for high throughput cell-based screening and
selection of specific sequences able to recognize
particular biomolecular targets [5-7]. We are using
this unique set of technologies for cell-based
screening and selection of genetically-encoded
libraries of cyclotides against particular protein
targets. Cyclotides
are ribosomally produced in plants from precursors
that comprise between one and three cyclotide
domains, however the mechanism of excision of the
cyclotide domains and ligation of the free N- and
C-termini to produce the circular peptides has not
been elucidated. Our group has recently developed
and successfully used a bio-mimetic approach for the
biosynthesis of folded cyclotides inside cells by
making use of modified protein splicing units (Fig.
2). Our important finding makes possible the
generation of large libraries of cyclotides (≈109)
for high throughput cell-based screening and
selection of specific sequences able to recognize
particular biomolecular targets [5-7]. We are using
this unique set of technologies for cell-based
screening and selection of genetically-encoded
libraries of cyclotides against particular protein
targets.
Cell-based screening: Available methods for
producing and screening high-affinity ligands
against particular molecular targets are either
based in rational or combinatorial approaches. The
rational approach usually requires the molecular
structure of the biomolecular target, and then
potential binders are selected from a virtual
library of compounds using docking software [8].
Despite recent advances in computing technology and
the development of adaptive docking software [8],
this is still a slow, although promising, process.
Combinatorial approaches, on the other hand, use
random generation of a large number of compounds
that are then screened against a biomolecular
target. Most of the methods for library screening,
however, are performed in vitro, which is a long and
laborious process. Cell-based screening, on the
other hand, opens the possibility of using single
cells as microfactories where the biosynthesis and
screening of particular ligands can take place in a
single process within the same cellular cytoplasm
[9]. The use of a complex molecular environment,
such as the cellular cytoplasm, provides the ideal
background to identify highly specific inhibitors.
Furthermore, the recent introduction of genetically
encoded fluorescence-based assays [10] allows the
use of high-throughput screening methods such as
fluorescence-activated cell sorting (FACS) to study
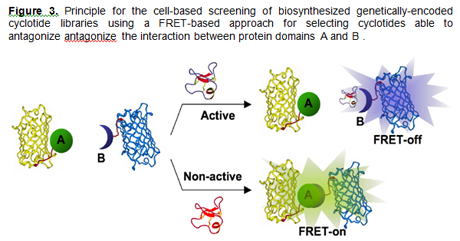
molecular interactions inside living cells [11]. Our
group has recently reported a cell-based screening
approach for Anthrax Lethal Factor antagonists [12].
This approach used the CyPet and YPet fluorescent
proteins as a FRET-couple to screen
genetically-encoded libraries of cyclotides inside
living bacterial cells [11-13] (Fig. 3). This
screening approach is optimized for use in E. coli
in combination with FACS, and it is designed to
screen large libraries meanwhile minimizing the
number of false positives.
 We
are combining this set of unique technologies for
finding specific de novo sequences of cyclotides
able to bind to particular serum proteins markers
for early detection of ovarian cancer as well as
inactivate some key interactions involved in tumor
cell proliferation and suppression. We
are combining this set of unique technologies for
finding specific de novo sequences of cyclotides
able to bind to particular serum proteins markers
for early detection of ovarian cancer as well as
inactivate some key interactions involved in tumor
cell proliferation and suppression.
Conclusions and outlook
Cyclotides are small globular micro proteins with a
unique head-to-tail cyclized backbone, which is
stabilized by three disulfide bonds [14]. The number
and positions of cysteine residues are conserved
throughout the family, forming the cyclic cystine-knot
motif (CCK) [14] that acts as a highly stable and
versatile scaffold on which hyper-variable loops are
arranged. This CCK framework gives the cyclotides
exceptional resistance to thermal and chemical
denaturation and enzymatic degradation. This is
particularly important for the selection of
protein-capture reagents able to work in
biologically complex samples such as tears, blood,
plasma and other biological fluids, which high
content in proteases. Together, these
characteristics make cyclotides ideal candidates to
be used as molecular scaffolds for the discovery of
stable high affinity ligands against particular
biomolecular targets, thus replacing the less stable
antibody-based scaffolds which have been
traditionally used as the protein capture reagents
of choice.
Dr. Julio A. Camarero is
Associate Professor at the Department of
Pharmaceutical Sciences in the University of
Southern California since 2008. He studied chemistry
at the University of Barcelona, received Masters
Degree in 1992 and completed his PhD Thesis in 1996.
Then he joined the group of Professor Tom W. Muir at
The Rockefeller University as a Burroughs Wellcome
Fellow where he contributed to the development of
new chemo selective ligation techniques for the
chemical engineering of proteins to study bacterial
transcription. In 2000, he moved to the Lawrence
Livermore National Laboratory as a Distinguished
Lawrence Fellow where he became staff scientist and
head of laboratory in 2003. He finally joined the
School of Pharmacy at the University of Southern
California in 2008 as Associate Professor. His
current research interests are focused in the
development of new bioorganic approaches using
protein splicing and synthetic protein chemistry for
studying biological processes involved in bacterial
pathogenicity and cancer and how can be modulated or
inhibited by highly constrained cyclic peptides. Dr.
Camarero has authored over 40 peer-reviewed
publications and four invited book chapters. For
article feedback, contact Dr. Camarero at
jcamarer@pharmacy.usc.edu
|
|
 |
|
